"T’i njohim sëmundjet e rralla": Paraplegjia e trashëguar spastike/ Sëmundja strampel–lorein
Shkup, 16 shkurt 2026
Në muajin me më pak ditë të vitit, në muajin që edhe këtë vit mbetet i veçantë me 28 ditët e tij të rralla, dhe në muajin të cilin pacientët me sëmundje të rralla e identifikojnë si të TYRIN – për të tetën herë me radhë do të shpalosim historitë e guximshme për t’ju njohur më afër me të gjithë ata që jetojnë me sfida dhe probleme të padukshme. Le të nisim një kapitull të ri mirëkuptimi dhe empatie, sepse së bashku zotërojmë fuqinë e bashkimit, forcën e shpresës, fuqinë e informacionit dhe guximin për t’u dhënë ngjyrë hijeve, duke sjellë dritë dhe dukshmëri...
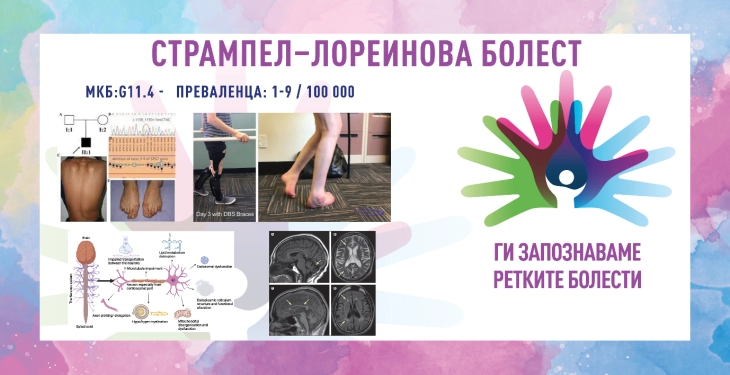
PARAPLEGJIA E TRASHËGUAR SPASTIKE/ SËMUNDJA STRAMPEL–LOREIN/ HEREDITARY SPASTIC PARAPLEGIA/ STRÜMPELL–LORRAIN DISEASE
ICD-10 G11.4
PREVALENCA – paraqitet te 1–9/100 000 banorë; në Evropë haset nga 1/11 000 deri në 1/77 000 banorë.
HYRJE
Paraplegjia e trashëguar spastike (HSP), e njohur gjithashtu si sëmundja Strampel-Lorein, është grup shumë heterogjen i sëmundjeve trashëguese, tipari kryesor i të cilave është çrregullim progresiv në ecje. Sëmundja manifestohet me ngurtësi progresive (spasticitet) dhe tkurrje të
ekstremiteteve të poshtme. Simptomat janë si rezultat i mosfunksionimit të aksoneve të gjata në palcën kurrizore. Qelizat e prekura janë neuronet motorike primare, kështu që është sëmundje e neuronit të sipërm motorik. Sëmundja shkaktohet nga defekte në transportin e proteinave, proteinave strukturore, proteinave të mirëmbajtjes së qelizave, lipideve dhe substancave të tjera në të gjithë qelizën. Fijet e gjata nervore (aksonet) preken sepse distancat e gjata i bëjnë qelizat nervore veçanërisht të ndjeshme ndaj defekteve në mekanizmat e përmendur. Fillon në fëmijëri ose në adoleshencën e hershme dhe përparon ngadalë gjatë gjithë jetës. Sëmundja u përshkrua për herë të parë në vitin 1880 nga neurologu gjerman Adolf Strümpel, ndërsa u përshkrua më gjerësisht në vitin 1888 nga mjeku francez Maurice Lorrain. Termi "paraplegji e trashëguar spastike" u mendua nga Anita Harding në vitin 1983.
ETIOLOGJIA
Sëmundja Strampel-Lorein ndodh për shkak të mosfunksionimit të neuroneve të sipërme motorike të traktit kortikospinal, i transmetuar në mënyrë të trashëguar, zakonisht në mënyrë autosomale dominante, por mund të jetë edhe autosomale recesive. Kjo do të thotë se sëmundja mund të trashëgohet nga njëri ose të dy prindërit, në varësi të llojit të mutacionit. Më së shpeshti shoqërohet me mutacione në gjenet SPAST (2p22.3), ATL1 (14q22.1), REEP1 (2p11.2) dhe KIF5A (12q.13.3) për HSP-të autosomale dominante dhe SPG7 (16q24.3), SPG11. (15q21) . 1), ndërsa CYP7B1 (8q12.3) për sëmundjen autosomale recesive, të cilat kodojnë proteina të rëndësishme për ruajtjen e funksionit normal të neuroneve motorike në palcën kurrizore. Deri më tani janë identifikuar më shumë se 80 gjene përgjegjëse për shfaqjen e sëmundjes. Proteinat e koduara janë të përfshira në shumë procese, duke përfshirë transportin aksonal, mielinizimin, shkëmbimin e endomembranës, funksionet mitokondriale, metabolizmin kompleks të lipideve dhe nukleotideve. Këto mutacione çojnë në degjenerim të neuroneve kortikospinale dhe dëmtim të rrugëve nervore spinale, duke ndërprerë transmetimin e nevojshëm për të kontrolluar lëvizjet e muskujve të gjymtyrëve të poshtme. Megjithatë, një numër i konsiderueshëm i pacientëve janë pa diagnozë gjenetike pas analizave sistematike.
SIMPTOMAT DHE DIAGNOSTIKIMI
Paraplegjia e trashëguar spastike manifestohet me përkeqësim progresiv të forcës së muskujve të këmbëve, si dhe rritje të spasticitetit. Simptomat kryesore përfshijnë:
§ spasticitetin – tashmë në fazën e hershme të sëmundjes, pacientët fillojnë të vërejnë tension të shtuar të muskujve dhe vështirësi në aktivitetet e lëvizjes, si ecja ose ngritja e këmbëve;
§ spasticitetin e ekstremiteteve të poshtme – dobësim gradual i muskujve të këmbëve, që çon në lëvizje të kufizuar ose nevojë për mjete ndihmëse për ecje;
§ efektet në koordinim dhe ekuilibër – ekuilibri i trupit është i çrregulluar, ndërsa pacientët mund të kenë vështirësi në ecje, gjë që çon në rënie të shpeshta;
§ dhimbjen e muskujve dhe kyçeve – dhimbja kronike mund të paraqitet për shkak të tensionit të rritur në muskuj;
§ përkeqësimin progresiv – sëmundja zakonisht ka një ecuri të ngadaltë progresive, por mund të ndryshojë në varësi të variantit specifik gjenetik dhe moshës së fillimit.
Klinikisht, paraplegjia e trashëguar spastike mund të ndahet në formën e pastër dhe formën e komplikuar (komplekse).
Forma e pastër e paraplegjisë së trashëguar spastike karakterizohet nga spasticiteti dhe dobësia progresive e ngadaltë e ekstremiteteve, zakonisht të shoqëruara me çrregullime urinare dhe anomali të thella sensore (ndjeshmëria e reduktuar ndaj dridhjeve të gjymtyrëve të poshtme).
Forma komplekse e paraplegjisë së trashëguar spastike dallohet nga prania ose mungesa e veçorive shtesë neurologjike. Karakteristikat neurologjike mund të përfshijnë çrregullime cerebelare (ataksi, nistagmus, tremor), neuropati periferike demielinizuese (çrregullime sensore dhe/ose motorike), çrregullime kognitive, defekte sensore, epilepsi, tipare miopatike, tipare ekstrapiramidale (parkinsonizëm, distoni), çrregullime psikiatrike, etj, të cilat mund të
jenë tregues të një nëntipi të caktuar gjenetik. Mungesa e manifestimeve neurologjike përfshin anomalitë okulistike (katarakt, retinit pigmentoz, degjenerim makular) dhe anomali ortopedike (skoliozë, dislokime të kyçeve dhe deformime të ndryshme të këmbëve).
Diagnoza bazohet në simptomat klinike, ekzaminimin neurologjik, ecurinë progresive të sëmundjes, matjet e biomarkerëve, MRI të trurit dhe shtyllës kurrizore (për të përjashtuar gjendjet e tjera të zakonshme neurologjike), historinë familjare, testimin gjenetik molekular dhe përjashtimin e diagnozave të tjera si: skleroza multipleks, anomalitë spinale vaskulare, mungesa e vitaminës B12, infeksioni me HTLV-I, skleroza anësore primare, paraliza cerebrale diplegjike, sëmundjet gjenetike metabolike, çrregullimi i akumulimit të metaleve në tru.
Konfirmimi përfundimtar i diagnozës mund të sigurohet vetëm duke kryer teste gjenetike që synojnë mutacionet e njohura gjenetike. Në veçanti, testimi gjenetik kërkohet në rastet kur një mutacion është identifikuar më parë në familje.
PASOJAT
Shumica e personave me paraplegji të trashëguar spastike kanë një jetëgjatësi normale. Simptomat e sëmundjes Strampel-Lorein përkeqësohen në mënyrë progresive me kalimin e kohës, gjë që mund të çojë në lëvizshmëri të kufizuar, rrezik të shtuar për lëndime dhe nevojë për përdorimin e mjeteve ndihmëse për të lëvizur. Ekzistojnë edhe komplikime, siç janë dhimbja kronike, deformimet e gjymtyrëve dhe pasojat psikosociale. Prognoza varet nga fenotipi, gjenotipi, si dhe nga vetë shprehja e gjenit.
Në disa raste, individët e prekur janë me aftësi të kufizuara të rënda, ndërsa në të tjera raste, individët e prekur janë në gjendje të kryejnë shumicën e aktiviteteve të përditshme pa pasur nevojë për ndihmë.
ТRAJTIMI
Aktualisht, nuk ka një trajtim specifik që do të parandalonte, ngadalësonte ose ndryshonte rrjedhën e sëmundjes. Terapia është simptomatike dhe trajtimet synojnë zbutjen e simptomave dhe përmirësimin e cilësisë së jetës nëpërmjet: fizioterapisë dhe rehabilitimit, trajtimit medikamentoz (barna kundër spasticitetit, analgjezikë për qetësimin e dhimbjeve), ndërhyrje kirurgjikale (në disa raste për korrigjimin e deformimeve), mbështetje psikologjike dhe të ngjajshme. Nga barnat rekomandohen:
§ baclofen - relaksues muskulor për relaksimin e muskujve dhe reduktimin e tonusit, i cili mund të administrohet në mënyrë orale ose intratekale;
§ diazepam dhe clonazepam - për të zvogëluar intensitetin e spazmave;
§ oxybutynin chloride dhe tolterodine tartrate - (agjentët antikolinergjikë) relaksues i pavullnetshëm i muskujve dhe agjent antispazmatik, që përdoren për të reduktuar spasticitetin e fshikëzës urinare te pacientët me probleme të kontrollit të fshikëzës urinare;
§ crosystem - për të reduktuar aktivitetin e shtuar të muskujve;
§ antidepresivë - për pacientët që përjetojnë depresion klinik.
Terapia fizikale është veçanërisht e rëndësishme për të ndihmuar rivendosjen dhe ruajtjen e aftësisë për të lëvizur, ajo gjithashtu ndihmon në uljen e tonusit të muskujve, për të ruajtur ose përmirësuar diapazonin e lëvizjeve dhe lëvizshmërinë, për të rritur forcën dhe koordinimin, për të parandaluar komplikimet, si nyjet e ngrira, kontraktimet ose plagët.
për projektin:
Shoqata e pacientëve Rrallë është të jesh i rrallë nga Ohri, edhe këtë shkurt, në kuadër të sezonit të tetë të projektit T’I NJOHIM SËMUNDJET E RRALLA 2026, ju njofton me sëmundjet e rralla nga të cilat vuan popullata në R.S. të Maqedonisë. Shpresojmë që përmes këtyre 28 diagnozave të reja të rralla, të gjithë së bashku do të arrijmë të ngrisim zërin për problemet me të cilat ballafaqohen personat me sëmundje të rralla, si dhe familjet e tyre.
Fushata është e karakterit informativ-edukativ dhe në të kanë punuar mbi 70 profesorë dhe mjekë eminentë nga Qendra Klinike “Nënë Tereza” në Shkup, mjekë nga institucionet shëndetësore të rajonit Ohër-Strugë – Spitali i Ohrit dhe i Strugës, ISHP Spitali Special për Ortopedi dhe Traumatologji “Shën Erazmo” - Ohër, ISHP Spitali i Specializuar për parandalimin, mjekimin dhe rehabilitimin e sëmundjeve kardiovaskulare “Shën Stefani” - Ohër, si dhe mjekë nga shumë ordina private nga komunat e R.S. të Maqedonisë. Ajo që vërtet shqetëson është numri i madh dhe diversiteti i sëmundjeve të rralla dhe informacionet për diagnozat vërtet ultra të rralla si: Fahr's Syndrome, Jalili syndrome, Marcus-Gunn syndrome, MoyaMoya disease, Shukla-Vernon syndrome...
Megjithatë, shpresa mbetet udhërrëfyesi ynë dhe sinqerisht besojmë se ky hulumtim do të na ndihmojë të krijojmë një bazë të dhënash që do të shërbejë për planifikimin e aktiviteteve të ardhshme në interes të personave me sëmundje të rralla, për lidhjen e familjeve dhe shkëmbimin e përvojave. Ajo që na inkurajon dhe na motivon të ecim përpara është efekti pozitiv i fushatave që kemi udhëhequr në vitet e kaluara. Jemi të vetëdijshëm se kemi ndihmuar dhe inkurajuar shumë pacientë dhe familje që të flasin publikisht për sëmundjen e tyre, të mbledhin guximin për të na kontaktuar, dhe natyrisht, kemi marrë shumë lëvdata nga komuniteti mjekësor profesional, i cili po përfshihet gjithnjë e më aktivisht në fushatat tona. Ju shprehim mirënjohje publike sepse pa ndihmën dhe këshillat e tyre profesionale nuk do t'ia dilnim dot.
Dita e parë e sëmundjeve të rralla u shënua në vitin 2008, saktësisht më 29 shkurt, një datë e RRALLË që ndodh vetëm një herë në çdo katër vjet. Që atëherë, Dita e Sëmundjeve të Rralla mbahet çdo vit në ditën e fundit të shkurtit, muaj i njohur për numrin e tij të RRALLË të ditëve. Dita e Sëmundjeve të Rralla është një fushatë ndërkombëtare për rritjen e vetëdijes. Nga fillimi në vitin 2008, mijëra ngjarje janë organizuar në mbarë botën duke përfshirë miliona njerëz. Fushata filloi si një ngjarje evropiane dhe me kalimin e kohës u bë fenomen global, me pjesëmarrjen e mbi 90 vendeve të botës në vitin 2018. R.S. e Maqedonisë u përfshi për herë të parë në shënimin e kësaj dite të rrallë në vitin 2012. Fushata për Ditën e Sëmundjeve të Rralla është për publikun e gjerë, për institucionet relevante dhe kushdo është i mirëpritur të bashkohet: pacientët dhe familjet, organizatat e pacientëve, profesionistët mjekësorë, studiuesit, industria farmaceutike, institucionet e shëndetit publik – sa më shumë të jemi, aq më mirë. Dita e Sëmundjeve të Rralla është një mundësi që pjesëmarrësit të jenë pjesë e thirrjes globale drejt politikëbërësve, studiuesve, kompanive dhe profesionistëve shëndetësorë për të përfshirë më shumë dhe më me efikasitet pacientët në kërkimet për sëmundjet e rralla.
BASHKOHUNI ME NE!











