"T’i njohim sëmundjet e rralla 2026": Sindroma e thyerjes Nijmegen/ Nijmegen breakage syndrome
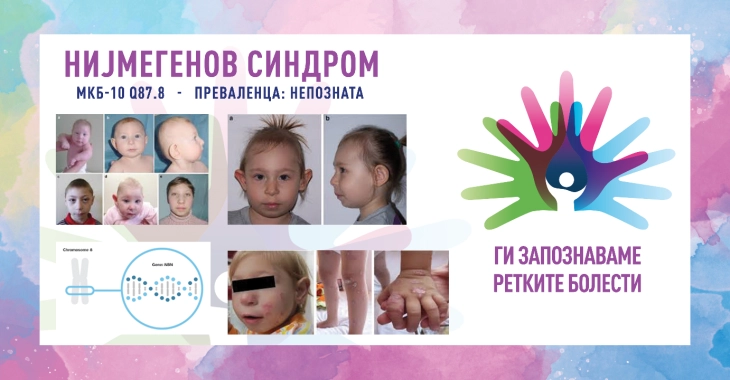
SINDROMA E THYERJES NIJMEGEN/ NIJMEGEN BREAKAGE SYNDROME
ICD-10 Q87.8 (Q90.9, D81.9)
PREVALENCA
– e panjohur, megjithëse ka të dhëna që prevalenca te të porsalindurit me origjinë sllave lindore (Bjellorusia, Ukraina, Rusia dhe Letonia) është vlerësuar në 1/1 000 000 lindje të gjalla, ndërsa prevalenca në rajonin sllav perëndimor (Poloni, Sllovaki dhe Republika Çeke) u vlerësua në 1/330 000.
HYRJE
Sindroma e thyerjes Nijmegen (NSR) është sindromë e rrallë e paqëndrueshmërisë gjenetike, kromozomale që ka shumë të ngjarë të jetë rezultat i një defekti në mekanizmin e riparimit të thyerjes së dyfishtë të ADN-së ose mekanizmit të pjekjes për riparimin e thyerjes së dyfishtë të ADN-së. Ky çrregullim autosomal recesiv u përshkrua për herë të parë në pacientët holandezë, të cilët me gjasë ishin pasardhës të qytetarëve polakë ose çekë që emigruan në Holandë gjatë viteve 1600 pas Luftës 30-vjeçare, sepse shumica e pacientëve me NSR jetojnë në Poloni dhe Republikën Çeke. Në lindje, foshnjat prezantohen me mikrocefali, tipare dismorfike të fytyrës që bëhen më të dukshme me moshën, ngadalësim të rritjes, infeksione të përsëritura sinopulmonare dhe një frekuencë jashtëzakonisht të lartë të sëmundjeve malinje në moshë të hershme. Modeli i trashëgimisë është autosomik recesiv.
ETIOLOGJIA
Sindroma e thyerjes Nijmegen shkaktohet nga mutacionet në gjenin NBN (8q21-q24), veçanërisht brenda ekzoneve 6-10, të cilat çojnë në fragmente të cunguara pjesërisht funksionale të nibrinës, një produkt gjeni i përfshirë në riparimin e thyerjeve të dyfishtë të ADN-së. Mbi 90% e pacientëve me origjinë sllave janë homozigotë.
SIMPTOMAT DHE DIAGNOSTIKIMI
Manifestimet klinike nuk janë tipike dhe mund të ndryshojnë në ashpërsi. Shenjat kryesore janë mikrocefalia e pranishme në lindje e cila përparon me kalimin e moshës, tiparet dismorfike të fytyrës (vija e mesme e theksuar e theksuar nga një ballë e përkulur dhe tërheqja e mandibulës), vonesa e lehtë e rritjes dhe në pacientët femra ndodh shpesh dështimi i parakohshëm i vezoreve. Janë raportuar një sërë anomalish kongjenitale, duke përfshirë në sistemin nervor qendror (hidrocefalus, skizencefali, kista arachnoid), traktin respirator (kuzë e çarë/qiellzë, atrezi choanale), sistemin urogjenital (veshka patkoi, veshka ektopike/distopike, hipospadia, kriptoria ), anomalitë e lehta skeletore (polidaktilia para dhe postaksiale, hipoplastike ose gishti i dyfishtë, klinodaktilia e gishtit të pestë). Shpesh shihen njolla kafeje ose vitiligo dhe disa pacientë zhvillojnë nerva të shumëfishtë të pigmentuar. Manifestime të tjera kryesore përfshijnë mungesën e imunitetit me infeksione të përsëritura të traktit respirator, një predispozitë të fortë ndaj tumoreve malinje (kryesisht limfoide, por shfaqen edhe tumore të ngurta) dhe ndjeshmëri ndaj radios. Deri në moshën 20 vjeçe, mbi 40% e pacientëve zhvillojnë një sëmundje malinje dhe janë të prirur për zhvillimin e tumoreve malinje dytësore. Zhvillimi kognitiv është afër mesatares (normale/kufitare) në fëmijëri dhe parashkollor, por aftësitë intelektuale bien gradualisht me kalimin e moshës (i butë deri në mesatar).
Diagnoza bazohet në manifestimet klinike dhe konfirmohet ose nga sekuenca me një gjen (zakonisht për popullatat sllave) ose nga panelet e sekuencës së gjeneratës së ardhshme multigjenike. Aty ku testimi gjenetik nuk është i disponueshëm, diagnoza mund të mbështetet nga dëshmitë e paqëndrueshmërisë kromozomale (spontane dhe të induktuar), ndjeshmërisë së rritur qelizore ndaj rrezatimit jonizues, in vitro, mungesës së imunitetit të kombinuar dhe mungesës së plotë të nibrinës me gjatësi të plotë. Historia familjare (malinje, mikrocefali ose hidrocefalus, vdekja e hershme e një vëllai ose motre) gjithashtu mund të përcaktojë diagnozën. Është veçanërisht e rëndësishme të përjashtohen diagnozat diferenciale si anemia Fanconi, sindroma LIG4, mungesa e Cernunnos-XLF, çrregullimi i ngjashëm me NBS, çrregullimi i ngjashëm me ataksi-teleangiektaze dhe sindroma e Bloom-it.
PASOJAT
Prognoza është e keqe, ku shkaku kryesor i vdekjes është malinjiteti. Pacientët që zhvilluan limfomën ose leukeminë vdiqën nga progresi i sëmundjes, relapsat dhe sëmundjet malinje sekondare. Efekti i dobishëm i HSCT në mbijetesën afatgjatë është konfirmuar në studimet e fundit.
TRAJTIMI
Fatkeqësisht për këtë syndrome si shumica e tyre nuk ka terapi specifike. Diagnoza e hershme ka një rëndësi të madhe për të shmangur infeksionet e rënda të përsëritura, ekspozimin e panevojshëm ndaj rrezatimit për qëllime diagnostikuese dhe efektet negative të radioterapisë së tumorit. Prindërit e fëmijës së prekur janë bartës të detyrueshëm të mutacioneve të NSC dhe kështu, për çdo shtatzëni, ekziston një rrezik 25% që pasardhësit të trashëgojnë sëmundjen. Bartësve të mutacionit themelues sllav duhet t'u ofrohet ndjekja e kancerit, veçanërisht e gjirit te femrat dhe e prostatës te meshkujt. Pacientët kërkojnë trajtim multidisiplinar dhe ndjekje afatgjatë (malinjiteti, imunodefiçenca, rritja, hipogonadizmi hipergonadotropik te gratë). Monitorimi i sistemit imunitar është jashtëzakonisht i rëndësishëm gjatë gjithë jetës; metodat dhe llojet specifike të imunizimit janë të nevojshme (rekomandohen vaksinat acelulare). Në rast malinje limfoide, transplantimi i qelizave staminale hematopoietike (HSCT) rekomandohet pas faljes së parë të plotë. Për femrat ofrohet monitorim nga mosha 12 vjeçe (endokrinolog/gjinekolog) dhe terapi hormonale e përshtatshme për moshën.
për projektin:
Shoqata e pacientëve Rrallë është të jesh i rrallë nga Ohri, edhe këtë shkurt, në kuadër të sezonit të tetë të projektit T’I NJOHIM SËMUNDJET E RRALLA 2026, ju njofton me sëmundjet e rralla nga të cilat vuan popullata në R.S. të Maqedonisë. Shpresojmë që përmes këtyre 28 diagnozave të reja të rralla, të gjithë së bashku do të arrijmë të ngrisim zërin për problemet me të cilat ballafaqohen personat me sëmundje të rralla, si dhe familjet e tyre.
Fushata është e karakterit informativ-edukativ dhe në të kanë punuar mbi 70 profesorë dhe mjekë eminentë nga Qendra Klinike “Nënë Tereza” në Shkup, mjekë nga institucionet shëndetësore të rajonit Ohër-Strugë – Spitali i Ohrit dhe i Strugës, ISHP Spitali Special për Ortopedi dhe Traumatologji “Shën Erazmo” - Ohër, ISHP Spitali i Specializuar për parandalimin, mjekimin dhe rehabilitimin e sëmundjeve kardiovaskulare “Shën Stefani” - Ohër, si dhe mjekë nga shumë ordina private nga komunat e R.S. të Maqedonisë. Ajo që vërtet shqetëson është numri i madh dhe diversiteti i sëmundjeve të rralla dhe informacionet për diagnozat vërtet ultra të rralla si: Fahr's Syndrome, Jalili syndrome, Marcus-Gunn syndrome, MoyaMoya disease, Shukla-Vernon syndrome...
Megjithatë, shpresa mbetet udhërrëfyesi ynë dhe sinqerisht besojmë se ky hulumtim do të na ndihmojë të krijojmë një bazë të dhënash që do të shërbejë për planifikimin e aktiviteteve të ardhshme në interes të personave me sëmundje të rralla, për lidhjen e familjeve dhe shkëmbimin e përvojave. Ajo që na inkurajon dhe na motivon të ecim përpara është efekti pozitiv i fushatave që kemi udhëhequr në vitet e kaluara. Jemi të vetëdijshëm se kemi ndihmuar dhe inkurajuar shumë pacientë dhe familje që të flasin publikisht për sëmundjen e tyre, të mbledhin guximin për të na kontaktuar, dhe natyrisht, kemi marrë shumë lëvdata nga
komuniteti mjekësor profesional, i cili po përfshihet gjithnjë e më aktivisht në fushatat tona. Ju shprehim mirënjohje publike sepse pa ndihmën dhe këshillat e tyre profesionale nuk do t'ia dilnim dot.
Dita e parë e sëmundjeve të rralla u shënua në vitin 2008, saktësisht më 29 shkurt, një datë e RRALLË që ndodh vetëm një herë në çdo katër vjet. Që atëherë, Dita e Sëmundjeve të Rralla mbahet çdo vit në ditën e fundit të shkurtit, muaj i njohur për numrin e tij të RRALLË të ditëve. Dita e Sëmundjeve të Rralla është një fushatë ndërkombëtare për rritjen e vetëdijes. Nga fillimi në vitin 2008, mijëra ngjarje janë organizuar në mbarë botën duke përfshirë miliona njerëz. Fushata filloi si një ngjarje evropiane dhe me kalimin e kohës u bë fenomen global, me pjesëmarrjen e mbi 90 vendeve të botës në vitin 2018. R.S. e Maqedonisë u përfshi për herë të parë në shënimin e kësaj dite të rrallë në vitin 2012. Fushata për Ditën e Sëmundjeve të Rralla është për publikun e gjerë, për institucionet relevante dhe kushdo është i mirëpritur të bashkohet: pacientët dhe familjet, organizatat e pacientëve, profesionistët mjekësorë, studiuesit, industria farmaceutike, institucionet e shëndetit publik – sa më shumë të jemi, aq më mirë. Dita e Sëmundjeve të Rralla është një mundësi që pjesëmarrësit të jenë pjesë e thirrjes globale drejt politikëbërësve, studiuesve, kompanive dhe profesionistëve shëndetësorë për të përfshirë më shumë dhe më me efikasitet pacientët në kërkimet për sëmundjet e rralla.











